
Myeloproliferative neoplasms (MPNs) are characterized by clonal expansion of abnormal hematopoietic stem cells, which involves quantitative and qualitative changes that switch these cells from a resting to a procoagulant phenotype. MPNs usually get worse over time because of the increasing number of cells that build up in the blood and bone marrow. MPNs may develop into acute myeloid leukemia, chronic myelogenous leukemia (CML), polycythemia vera, primary myelofibrosis, essential thrombocythemia, chronic neutrophilic leukemia, and chronic eosinophilic leukemia. CML can lead to thrombotic and hemorrhagic complications and can transform into myelofibrosis and acute leukemia [1].
Relevant mutations
Mutations in JAK2, MPL, and CALR are phenotypic driver mutations; the mutated gene expression in cell lines causes cytokine independent or hypersensitive growth, which also occurs in primary cells from MPN patients. At diagnosis, the clinical phenotype varies according to the JAK2, MPL, and CALR mutations, influencing clinical outcomes and the probability of thrombosis. MPN patients with JAK2 and MPL mutations have a higher risk of thrombosis and a worse prognosis than patients with CALR mutations; CALR type I mutations correspond to a better prognosis than CALR type II mutations. In addition, other mutations in epigenetic genes, such as TET2, DNMT3A, IDH1/2, EZH2, and ASXL have been found to be associated with the progression of these neoplasms [2].
Thrombosis pathogenesis in MPN patients
Thrombosis in MPN patients has complex pathogenesis. Disease-related factors and, more importantly, the presence of certain mutations can interact with non-disease patient-related factors (age, previous history of thrombotic events, obesity, hypertension, hyperlipidemia, genetic traits) and combined oral contraceptive pills, or estrogen-containing hormone replacement therapy, resulting in an increased risk of thrombosis [1].
Prothrombotic features include the expression by blood cells of procoagulant and proteolytic properties, the secretion of inflammatory cytokines, and the expression of adhesion molecules. Prothrombotic changes occur in the normal vascular endothelium in response to inflammatory cytokines, hyper-viscosity, and leukocyte-derived proteases. Endothelial adhesion receptor upregulation induces attachment of platelets, erythrocytes, and leukocytes to the vascular wall, leading to clotting reactions and fibrin deposition. MPN patients present a procoagulant background with a hypercoagulable state, a subclinical condition characterized by alterations in plasma thrombotic markers. High circulating procoagulant microparticle levels and acquired activated protein C resistance are also common hypercoagulability features in MPN patients [1].
There are four principal hemostatic marker alterations in MPN patients’ blood and vascular cells, further increasing in patients carrying the JAK2 mutation (Fig.1).
-
-
- Platelets circulate in an activated state, which provides a catalytic surface for the generation of thrombin that further amplifies their activation.
- Elevated hematocrit levels can increase the thrombotic risk by multiple pathways. Hyper-viscosity plays a major thrombotic role, and an increasing red cell mass displaces platelets toward the vessel wall, facilitating shear-induced platelet activation and enhancing platelet–platelet interactions.
- Neutrophils can activate the blood coagulation system, releasing proteolytic enzymes and reactive oxygen species and increasing the expression of CD11b on their surface, which can activate or damage platelets and endothelial cells.
- Reactive oxygen species and intracellular proteases released by activated neutrophils can detach or lyse endothelial cells, affecting thromboregulation. MPN patients present high levels of circulating endothelial cells [3]
-
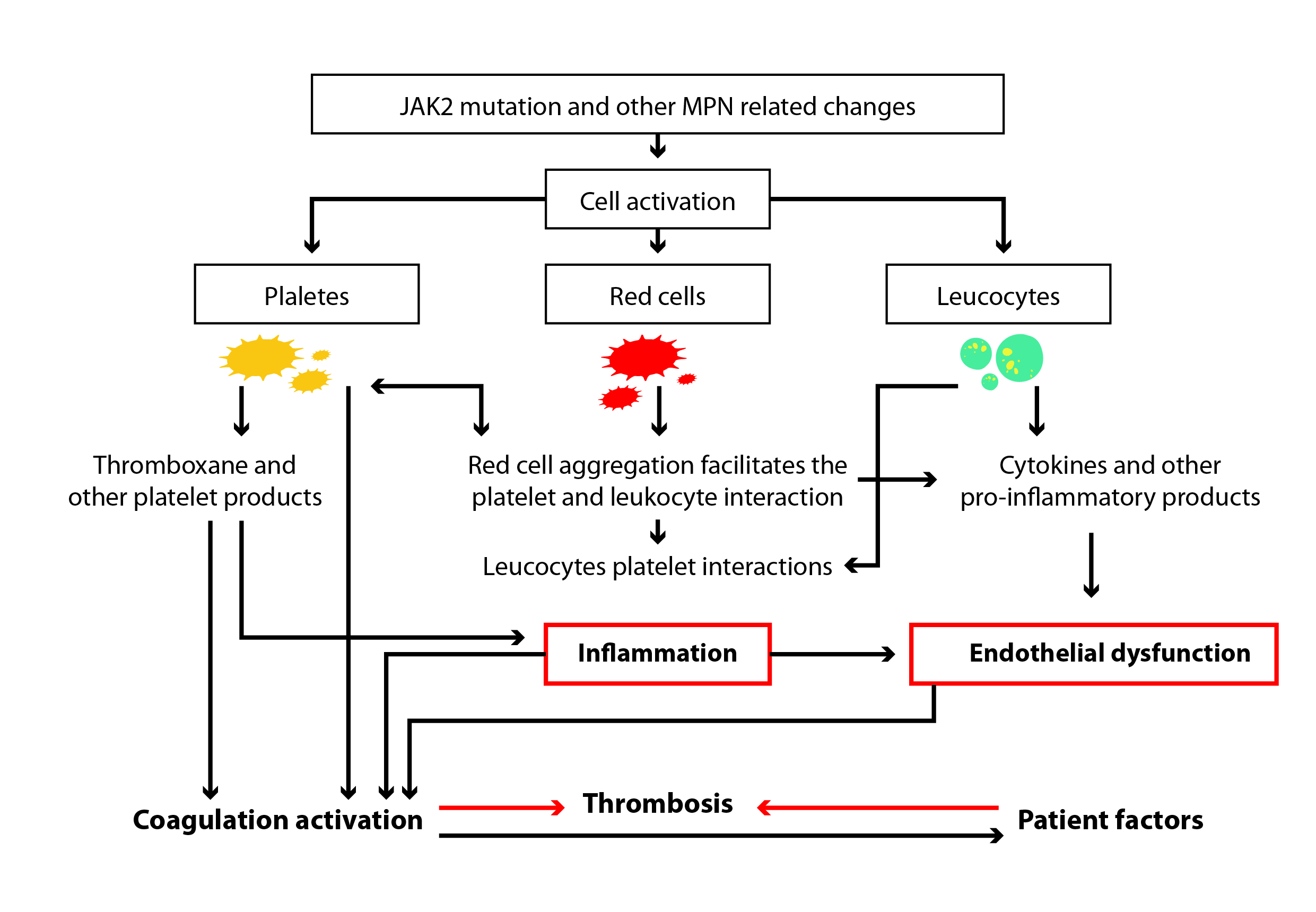
Figure 1: Mechanisms that, in myeloproliferative neoplasms, can increase the thrombotic risk through cell activation and inflammation [2].
MPN-associated thrombosis can happen both in the venous and arterial systems, but particularly in the cerebral venous, portal vein, mesenteric and splanchnic vein. If one of these symptoms present without obvious local pathology, molecular abnormalities associated with MPN should be investigated. Some patients develop thrombosis, particularly splanchnic vein thrombosis, associated with the JAK2 mutation but with a completely normal full blood count. Isolated JAK2 mutations occur in approximately 0.1–0.2% of the general population, and a myeloproliferative phenotype can develop in individuals bearing this allele over 4 or 5 years. Patients presenting with thrombosis at more usual sites, such as deep vein thrombosis and pulmonary embolism or arterial thrombosis, should be investigated for an underlying MPN if they have elevated hemoglobin or hematocrit (hemoglobin >165 g/L in men and >160 g/L in women, or hematocrit >0.49 in men and >0.48 in women) or a platelet count >450 × 109/L, as defined by the revised 2016 World Health Organization (WHO) criteria [4].
Tyrosine kinase inhibitors
Treatment for MPN depends on the type of the neoplasm. Tyrosine kinase inhibitors (TKIs), which target the Abl kinase domain, are the mainstay of the management of CML. However, three of the five BCR-ABL TKIs (dasatinib, nilotinib and ponatinib) increase the risk of developing arterial thrombosis. A recent study explored the mechanisms by which nilotinib and ponatinib promote acute and chronic thrombo-occlusion and the potential role of calcium channel blockade in facilitating these events. The results suggest that ponatinib induces a prothrombotic and proinflammatory phenotype in chronically treated CML patients and in acute C57BL/6 mice models. The thrombo-inflammatory phenotype induced by the TKIs ponatinib and nilotinib can be reversed with a calcium channel blocker, diltiazem, supporting the idea that ponatinib and nilotinib may exert a calcium influx effect contributing to endothelial/platelet activation and dysregulation.
As previously mentioned, platelets play a key role in arterial thrombotic events; the study showed that ponatinib activates endothelium and platelets in vitro, ex vivo, and in vivo. In addition, it potentiates platelet adhesion and thrombus formation in human and murine models. This prothrombotic effect was not observed with imatinib and dasatinib, which inhibited thrombus growth and stability over time. Furthermore, in an ex-vivo murine model, ponatinib exposure caused increased levels of pro-inflammatory markers (TNF-α, IFN-γ, and IL-6) and of soluble platelet selectin, a known marker of platelet and endothelium activation, predominantly derived from platelets. It seems that ponatinib and nilotinib TKIs modulate the release of arachidonic acid from membrane phospholipids and the synthesis and release of the vasoconstrictor TxA2 that is regulated by the movement of calcium ions. Plasma TxB2 levels were significantly increased in ponatinib- and nilotinib-treated CML patients but not in imatinib–dasatinib-treated or healthy controls.
In summary, the study suggests that nilotinib and ponatinib may have both acute and chronic prothrombotic effects. Chronic prothrombotic effects reflect a ‘thrombo-inflammatory’ phenotype involving platelet and endothelial activation and TxA2 generation [5].
Keep reading the ASH series:
MicroRNA and role in cancer-associated thrombosis
Interplay between the hematologic system and solid tumor progression
Late-breaking research presentations
References
- Arachchillage DR, Laffan M. Pathogenesis and management of thrombotic disease in myeloproliferative neoplasms. Semin Thromb Hemost. 2019;45(6):604-611. doi:10.1055/s-0039-1693477
- Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res. 2016;140 Suppl 1:S71-S75. doi:10.1016/S0049-3848(16)30102-5
- Barbui T, Finazzi G, Falanga A; Myeloproliferative neoplasms and thrombosis. Blood 2013;122(13):2176–2184. doi:1182/blood-2013-03-460154
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. doi:10.1182/blood-2016-03-643544
- Hamadi A, Grigg AP, Dobie G, et al. Ponatinib tyrosine kinase inhibitor induces a thromboinflammatory response. Thromb Haemost. 2019;119(7):1112-1123. doi:10.1055/s-0039-1688787